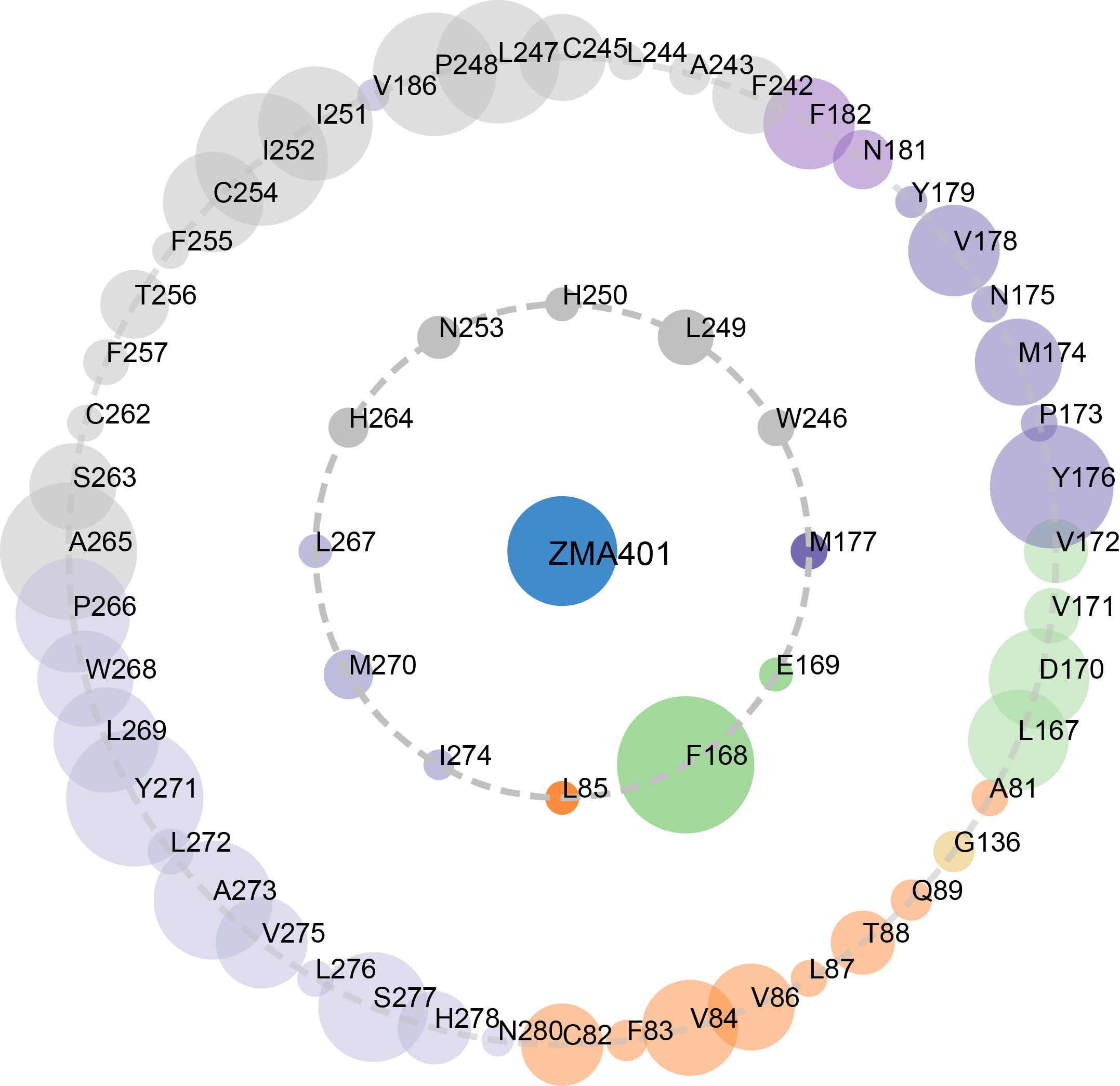
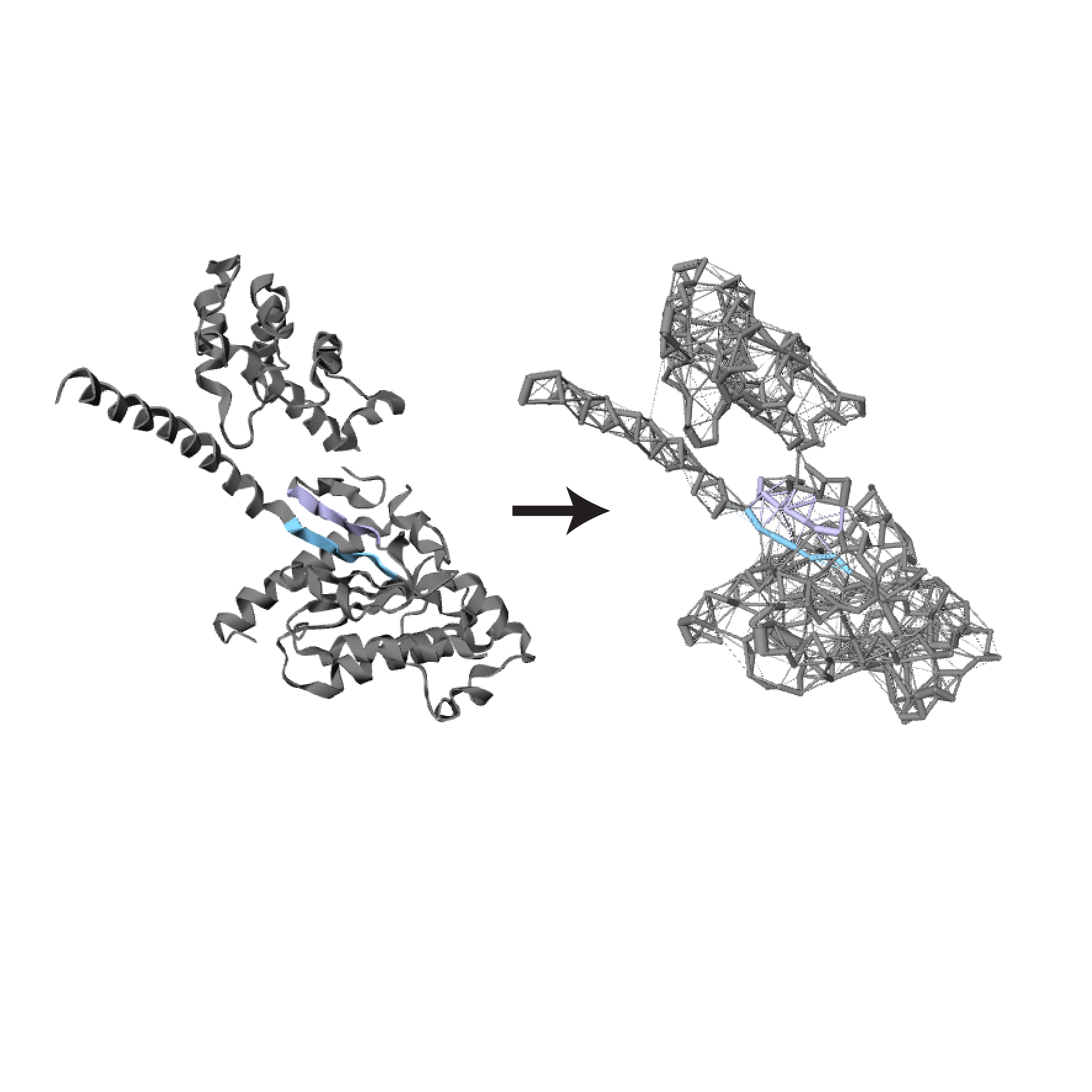
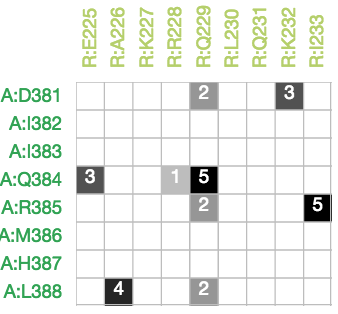
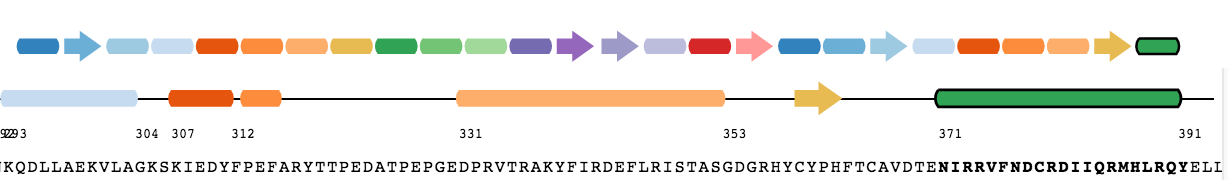
In this filtering step, contacts are normalized with respect to the the size of the amino acid. For the normalization, we used a previously published approach (Kannan et al, 1999 and Chakrabarty et al, 2016).
Briefly, we first identified all non-redundant crystal structures in the PDB (19.06.2017) from the NCBI database (ftp://ftp.ncbi.nih.gov/mmdb/ nrtable/) using a resolution cut-off of 2 Å. This resulted in 48,856 structures (95,159 chains).
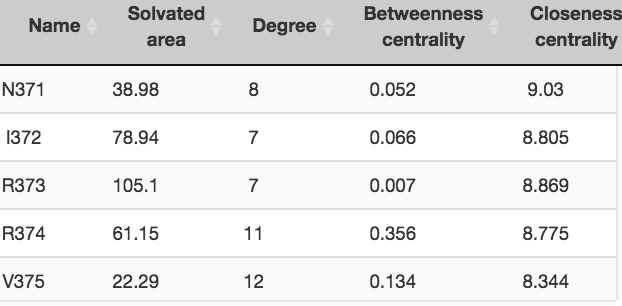
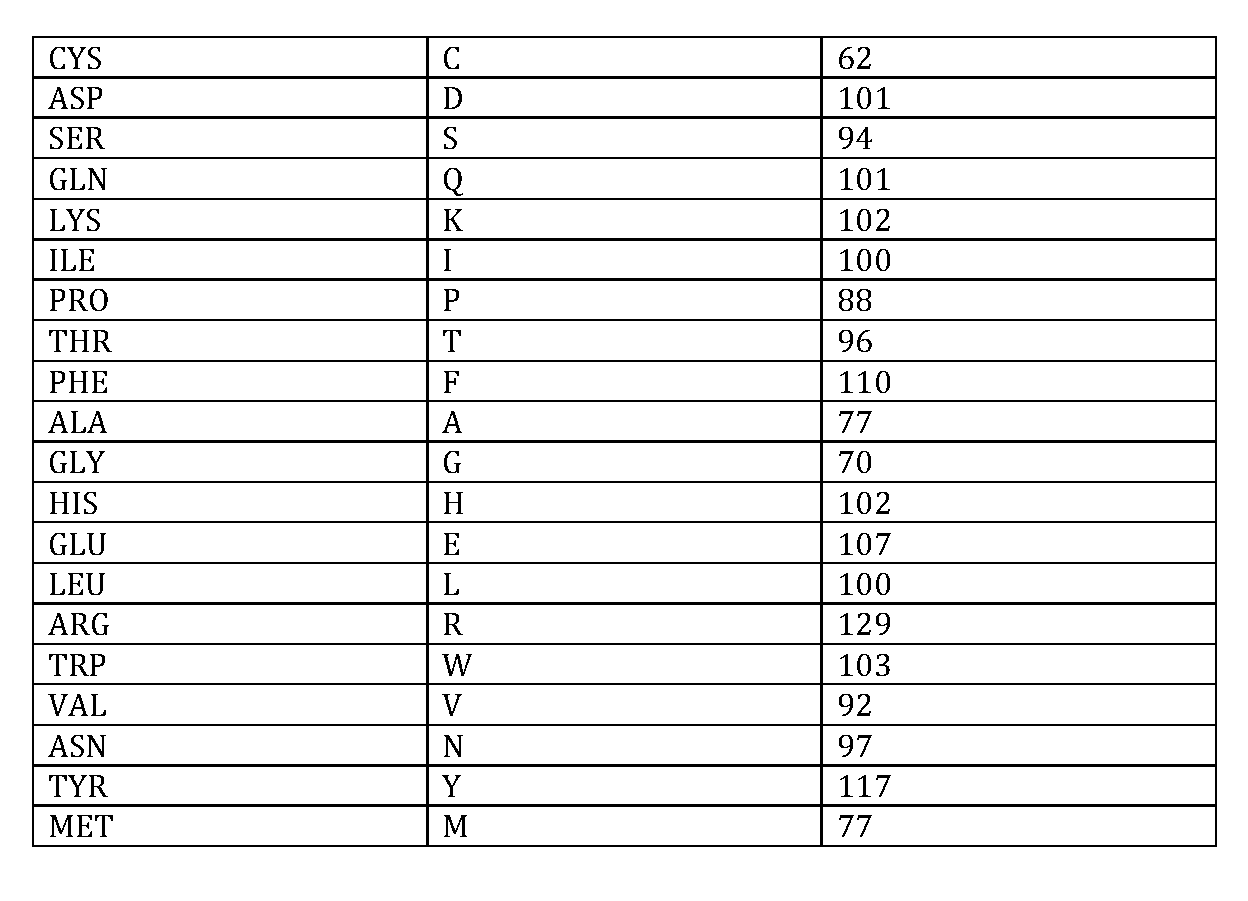
We then calculated the average of the maximum number of atomic contacts made by each of the 20 amino acids in these structures (see
table below). This was done using the precomputed results available as JSON files in Protein Contacts Atlas. Using this as our reference table,
we computed normalized contacts for any structure provided by the user using the following formula:
Normalized weight = (number of side chain atomic contacts /sqrt(norm_res1 * norm_res2))*100
where number of side chain atomic contacts is the number of atomic contacts between the side chains of two residues where the distance between two atoms is smaller than 4 Å and norm_res1 and norm_res2 are the values taken from the calculated table. After the normalized weight between the residues is calculated, any interaction which has the normalized weight smaller than the threshold chosen by the user is filtered.
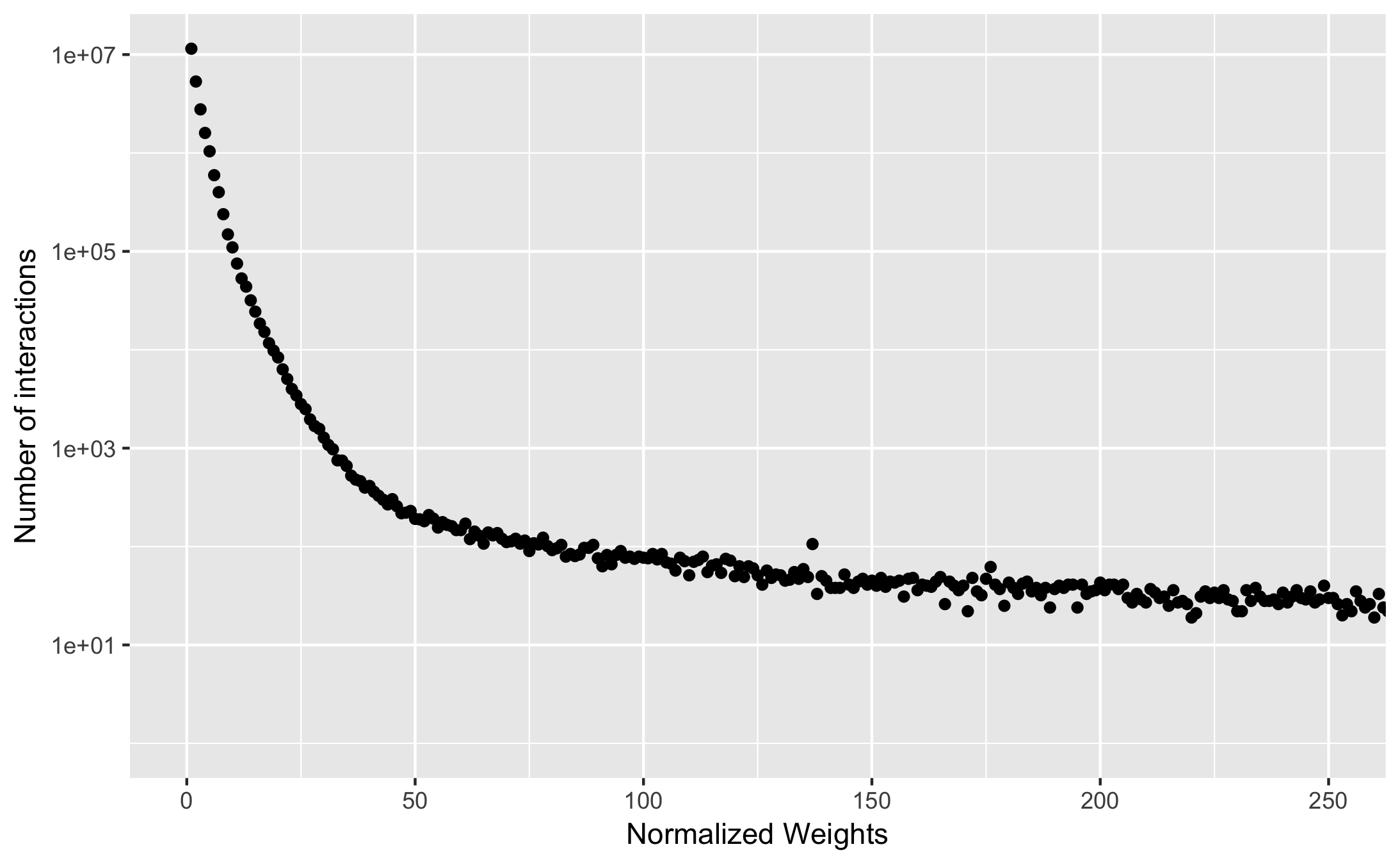
The table below shows a distribution of the interaction weights across all non-redundant structures in the PDB.
The user can choose a specific “normalized weight” cut-off based on the following table.
x- axis shows the normalized weights calculates and y-axis shows the total number of interactions with the associated normalized weights.