About
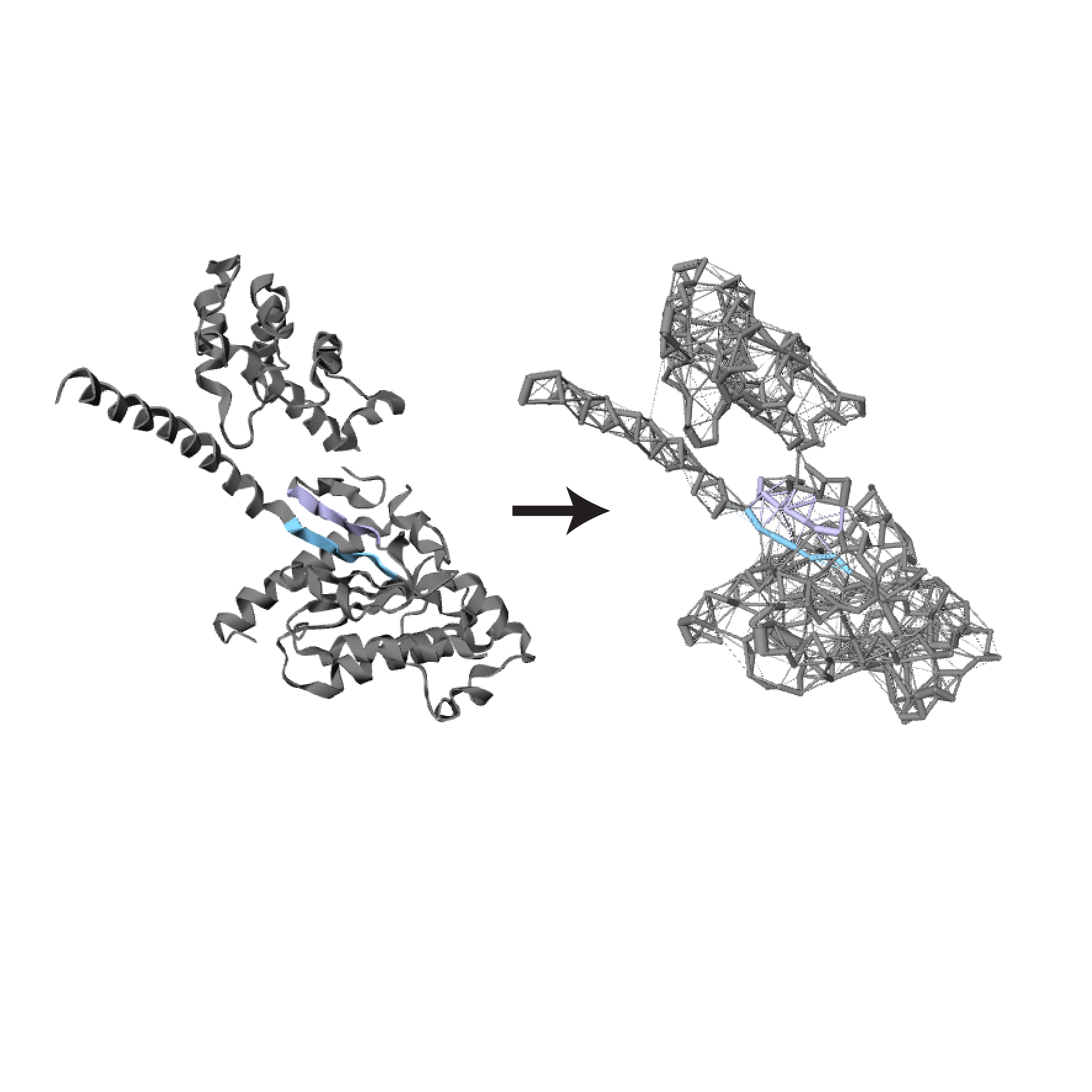
Protein Contacts Atlas is a tool that helps biologists analyze and visualize protein structures at atomic resolution using residue-residue interaction networks. It provides a range of representations, and can visualize interactions within a single protein or protein complex, or between a protein and nucleic acids, ligands or other small molecules. Residues can be classified based on sequence properties such as conservation, biophysical properties such as solvent accessible area, biochemical properties such as post-translational modifications (PTMs), or network properties such as closeness or betweenness of residue-residue interactions. Protein Contacts Atlas is also a database containing pre-computed networks for all crystal structures experimentally or computationally determined so far. It is a comprehensive resource for structural and molecular biologists analyzing either newly solved structures or previously solved structures deposited in the Protein Data Bank. Currently, the Protein Contacts Atlas database includes around 104,000 X-ray structures, and further structures will be automatically imported as they are deposited in the PDB. Protein Contacts Atlas combines a set of features that make it convenient and efficient to analyze structural data without requiring extensive programming knowledge. It is convenient to use as it contains preprocessed data for all structures in the PDB, it provides visualizations that are intuitive even for the non-expert, and it enables data and graphics to be easily exported.
How are the Residue Interaction Networks generated?
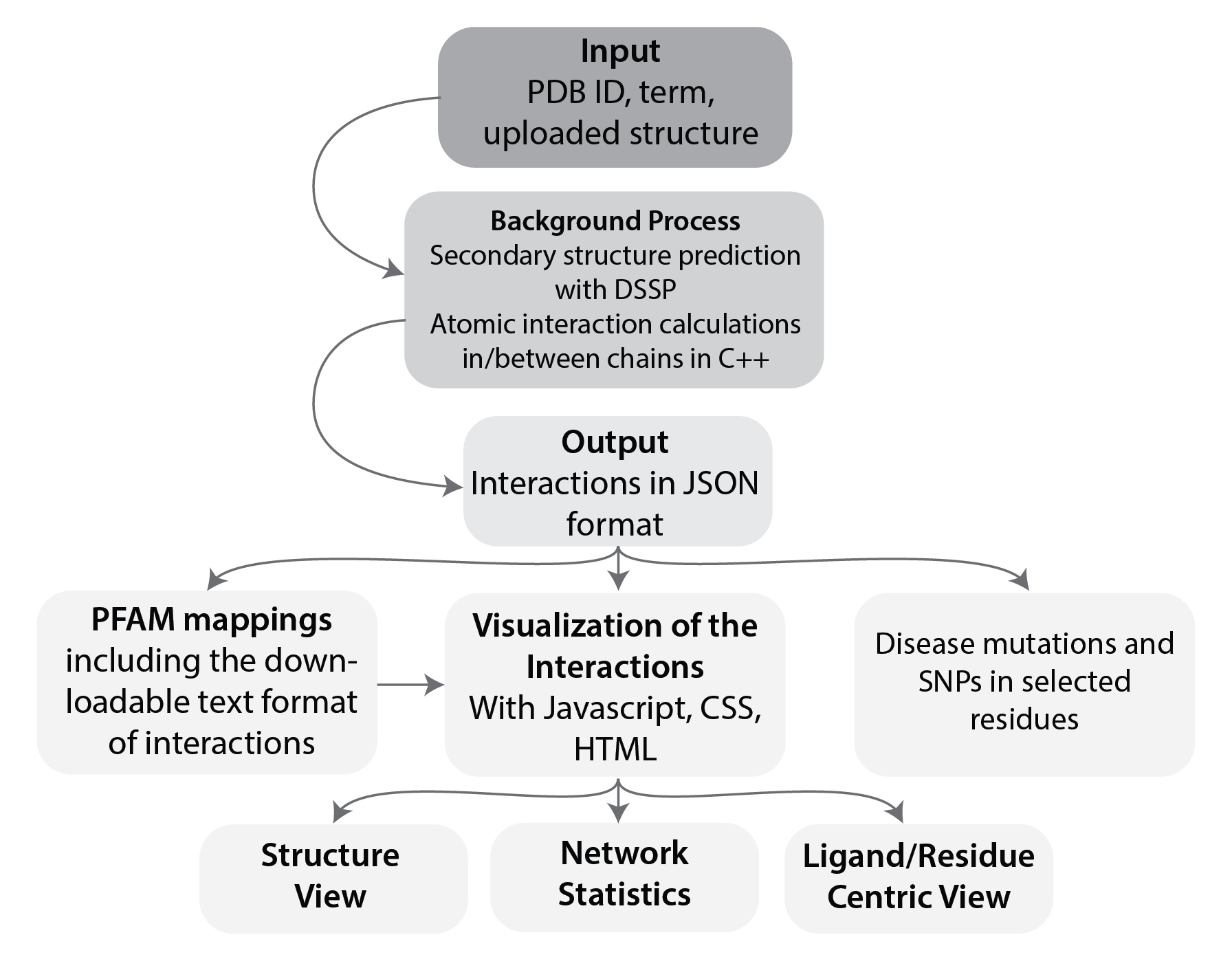
New structures added to the PDB are preprocessed as a batch process run monthly. Structures uploaded by a user undergo the same preprocessing steps, but at the time of uploading; depending on the size of the file, this may take 5-7 minutes. First, secondary structure elements are determined using DSSP. User-provided PDB files with just the atomic coordinates, without any additional information, is sufficient to perform data preprocessing. The atomic interaction calculations are performed by a custom-written C++ program. The coordinates of each atom are taken from the protein database (PDB) file and then the distance between every two atoms is calculated using Pythagoras' theorem.
Then, the Chothia radii for each residue and atom in these residues are taken. The sum of each of the atoms’ radii is subtracted from the distance and an interaction is assigned if the resulting distance is less than a threshold. By default, this threshold is 0.5 Å, but the user can choose any value in the range 0.5 to 0.8 Å with which to redo the calculations. In the case of heteroatoms, such as ligands, where there are no radii sizes recorded, an interaction is assigned if the distance is < 4 Å. The solvent accessible area for each residue is calculated using an external program called POPS. Results of pre-processing are stored in JSON files that are used to produce the interactive visualizations.
Information on downloadable files
Protein Contacts Atlas allows the users to download the residue interactions in text format for all chains per structure.
A zip file containing all the text files of a particular structure is downloaded. This link is accessible in the selected structure main page as well as
in "Advanced Features" section where the download can be done in batches for all PFAM domains.
The selected JSON files can be accessed via the Protein Contacts Atlas API which can be found here. The user adds the PDB ID and the chain on the link, here it is 3SN6 and chain A.
You can access quickly for a known structure's Protein Contacts Atlas interaction visualization page via the link http://www.mrc-lmb.cam.ac.uk/pca/redirect/PDBID where you change the PDBID (an example which shows information for structure
3SN6).
Protein Contacts Atlas also provides sif files to be used in Cytoscape. These can also be found on the main interaction page of Protein Contacts Atlas and can be downloaded for all chains as zip format.
Comparison of residue interactions
Protein Contacts Atlas does not provide visualisation for the comparison of interactions however we provide the interaction
files which can be downloaded individually for each structure. These files have the extension ".RIN" and has the basic information
of each residue interaction and how many atomic contacts every two residue has with each other. We recommend using ProSmart as a structure alignment tool,
written by Rob Nicholls in Garib Murshudov's group in MRC, Laboratory of Molecular Biology. The tool generates a common key for each residue in the structures selected to be aligned.
You can then combine this information with Protein Contacts Atlas's interaction files to do your comparisons.
Incorporating the visualisation of comparisons will be done in the next version of the server. For now, we recommend the users to use additional tabs for specific features and their comparisons.
Protein Contacts Atlas's Chord Plot is featured in PDBe
With an amazing work with our PDBe collaborators, we integrated the Chord plot feature of Protein Contacts Atlas in their website.
The link to their webpages can be found here.
Also, any developer who would like to use different components from PDBe including our Chord Plot component can go here where there is
a detailed description of how to integrate these components in their own websites. Chord plot can be found at the bottom of the page.
Arpeggio is used to compute different contact types
Protein Contacts Atlas can also calculate hydrogen bonds, water mediated hydrogen bonds, weak hydrogen bonds, ligand and metal complex interactions, salt bridges,
hydrophobic interactions, cation-pi interactions, pi-pi interactions, and other non-canonical contacts using Arpeggio (Jubb et al 2017). The link to their webpage can be found here.
The python source code of Arpeggio can be found here.